MOsaic CHromosomal Alterations (MoChA) Caller
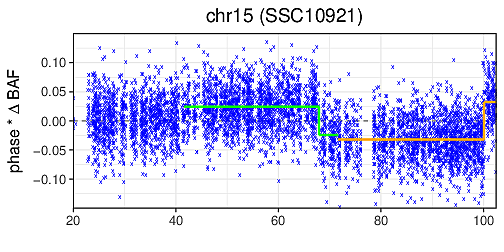 A computational tool to detect both germline and mosaic chromosomal alterations at low allelic fractions based on phased haplotype data. Mosaic chromsomal alterations in diploid cells affect the overall balance of alleles from the two haplotypes at consecutive heterozygous sites. Long-range phase information allows the careful integration of this imbalance across long chromosomal segments. By carefully performing this integration, MoChA is capable of detecting mosaic chromosomal alterations present in as little as one percent of the cells that comprise the DNA library. The ability to identify events at such low cell fractions greatly enhances the ability to detect clonality in samples from blood, cancer, and cell lines. MoChA is entirely written in C and based on HTSlib and BCFtools libraries and is equally capable of processing data from DNA microarray and whole-genome sequencing experiments, providing a simple unifying framework for researchers. Official Website Paper: PDF
A computational tool to detect both germline and mosaic chromosomal alterations at low allelic fractions based on phased haplotype data. Mosaic chromsomal alterations in diploid cells affect the overall balance of alleles from the two haplotypes at consecutive heterozygous sites. Long-range phase information allows the careful integration of this imbalance across long chromosomal segments. By carefully performing this integration, MoChA is capable of detecting mosaic chromosomal alterations present in as little as one percent of the cells that comprise the DNA library. The ability to identify events at such low cell fractions greatly enhances the ability to detect clonality in samples from blood, cancer, and cell lines. MoChA is entirely written in C and based on HTSlib and BCFtools libraries and is equally capable of processing data from DNA microarray and whole-genome sequencing experiments, providing a simple unifying framework for researchers. Official Website Paper: PDF
Mouse Brain Atlas
 The mammalian brain is composed of diverse, specialized cell populations. To systematically ascertain and learn from these cellular specializations, we used Drop-seq to profile RNA expression in 690,000 individual cells sampled from 9 regions of the adult mouse brain. We identified 565 transcriptionally distinct groups of cells using computational approaches developed to distinguish biological from technical signals. Cross-region analysis of these 565 cell populations revealed features of brain organization, including a gene-expression module for synthesizing axonal and presynaptic components, patterns in the co-deployment of voltage-gated ion channels, functional distinctions among the cells of the vasculature and specialization of glutamatergic neurons across cortical regions. Systematic neuronal classifications for two complex basal ganglia nuclei and the striatum revealed a rare population of spiny projection neurons. This adult mouse brain cell atlas, accessible through interactive online software (DropViz), serves as a reference for development, disease, and evolution. Interactive website to explore the atlas: http://dropviz.org/ Paper: PDF
The mammalian brain is composed of diverse, specialized cell populations. To systematically ascertain and learn from these cellular specializations, we used Drop-seq to profile RNA expression in 690,000 individual cells sampled from 9 regions of the adult mouse brain. We identified 565 transcriptionally distinct groups of cells using computational approaches developed to distinguish biological from technical signals. Cross-region analysis of these 565 cell populations revealed features of brain organization, including a gene-expression module for synthesizing axonal and presynaptic components, patterns in the co-deployment of voltage-gated ion channels, functional distinctions among the cells of the vasculature and specialization of glutamatergic neurons across cortical regions. Systematic neuronal classifications for two complex basal ganglia nuclei and the striatum revealed a rare population of spiny projection neurons. This adult mouse brain cell atlas, accessible through interactive online software (DropViz), serves as a reference for development, disease, and evolution. Interactive website to explore the atlas: http://dropviz.org/ Paper: PDF
Studying C4
 The complex variation of the complement component 4 (C4) genes has prevented their effective inclusion in genome-wide studies based on SNP arrays and exome sequencing. Here we provide resources to facilitate the analysis of C4 in both C4-focused and genome-wide studies.
The complex variation of the complement component 4 (C4) genes has prevented their effective inclusion in genome-wide studies based on SNP arrays and exome sequencing. Here we provide resources to facilitate the analysis of C4 in both C4-focused and genome-wide studies.
We describe a combination of molecular assays and downstream inferential strategies that utilize droplet digital PCR (ddPCR) to infer the C4 gene contents of each genome analyzed – including the combination of C4AL, C4AS, C4BL, and C4BS genes present. This method has high (64/64) concordance with results obtained using Southern blotting. Common C4 alleles can also be imputed from flanking SNPs. Though recurring mutation at C4 makes imputation less effective than it is for simpler variants, we found that the common C4 alleles can generally be imputed with 0.7 < r2 < 1, making this approach useful for large cohort studies. We provide the reference panel for imputation that we created from the HapMap samples.
Molecular analysis of C4 structural variation using droplet digital PCR
Studying Haptoglobin
 One of the first protein polymorphisms identified in humans involves the abundant blood protein haptoglobin. Two exons of the HP gene (encoding haptoglobin) exhibit copy number variation that affects HP protein structure and multimerization. The evolutionary origins and medical relevance of this polymorphism have been uncertain. We showed that this variation has likely arisen from many recurring deletions, more specifically, reversions of an ancient hominin-specific duplication of these exons. Although this polymorphism has been largely invisible to genome-wide genetic studies thus far, we describe a way to analyze it by imputation from SNP haplotypes and find among 22,288 individuals that these HP exonic deletions associate with reduced LDL and total cholesterol levels. We further show that these deletions, and a SNP that affects HP expression, appear to drive the strong association of cholesterol levels with SNPs near HP. Recurring exonic deletions in HP likely enhance human health by lowering cholesterol levels in the blood. Haptoglobin reference panel Paper: PDF
One of the first protein polymorphisms identified in humans involves the abundant blood protein haptoglobin. Two exons of the HP gene (encoding haptoglobin) exhibit copy number variation that affects HP protein structure and multimerization. The evolutionary origins and medical relevance of this polymorphism have been uncertain. We showed that this variation has likely arisen from many recurring deletions, more specifically, reversions of an ancient hominin-specific duplication of these exons. Although this polymorphism has been largely invisible to genome-wide genetic studies thus far, we describe a way to analyze it by imputation from SNP haplotypes and find among 22,288 individuals that these HP exonic deletions associate with reduced LDL and total cholesterol levels. We further show that these deletions, and a SNP that affects HP expression, appear to drive the strong association of cholesterol levels with SNPs near HP. Recurring exonic deletions in HP likely enhance human health by lowering cholesterol levels in the blood. Haptoglobin reference panel Paper: PDF
Studying the 17q21.32 Inversion
 Human chromosome 17q21.31 contains a megabase-long inversion polymorphism, many uncharacterized copy-number variations (CNVs) and markers that associate with female fertility, female meiotic recombination and neurological disease. Additionally, the inverted H2 form of 17q21.31 seems to be positively selected in Europeans. We developed a population genetics approach to analyze complex genome structures and identified nine segregating structural forms of 17q21.31. Both the H1 and H2 forms of the 17q21.31 inversion polymorphism contain independently derived, partial duplications of the KANSL1 gene; these duplications, which produce novel KANSL1 transcripts, have both recently risen to high allele frequencies (26% and 19%) in Europeans. An older H2 form lacking such a duplication is present at low frequency in European and central African hunter-gatherer populations. We further showed that complex genome structures can be analyzed by imputation from SNPs. The protocol generates 4 reference panels, two for HapMap and 1000 Genomes for GRCh37 and two for GRCh38. Protocol for reference panels Paper: PDF
Human chromosome 17q21.31 contains a megabase-long inversion polymorphism, many uncharacterized copy-number variations (CNVs) and markers that associate with female fertility, female meiotic recombination and neurological disease. Additionally, the inverted H2 form of 17q21.31 seems to be positively selected in Europeans. We developed a population genetics approach to analyze complex genome structures and identified nine segregating structural forms of 17q21.31. Both the H1 and H2 forms of the 17q21.31 inversion polymorphism contain independently derived, partial duplications of the KANSL1 gene; these duplications, which produce novel KANSL1 transcripts, have both recently risen to high allele frequencies (26% and 19%) in Europeans. An older H2 form lacking such a duplication is present at low frequency in European and central African hunter-gatherer populations. We further showed that complex genome structures can be analyzed by imputation from SNPs. The protocol generates 4 reference panels, two for HapMap and 1000 Genomes for GRCh37 and two for GRCh38. Protocol for reference panels Paper: PDF
Genome STRiP

Genome STRiP (Genome STRucture In Populations) is a set of software tools for analyzing genome structural variation in whole-genome sequence data from many individuals of the same species. Bob Handsaker in our lab is the lead developer and architect. We describe Genome STRiP and its applications in two papers in Nature Genetics. As of February 2015, Genome STRiP had been downloaded by 1,099 users and cited in 133 academic papers. Here are links to software downloads, documentation, cookbooks, and Bob Handsaker’s workshop videos for users. This work is supported by the iSeqTools program of the National Human Genome Research Institute. Bob Handsaker makes available his new maps of human genome copy number variation in the 1000 Genomes Project. These maps contain the genomic locations, alleles, and allele frequencies of 8,659 segregating CNVs in diverse populations, including 1,356 multi-allelic CNVs that are described for the first time at the levels of integer copy numbers, copy-number alleles, and allele frequencies. These maps also report the relationships of CNV alleles to SNPs and haplotypes, including visual plots suitable for re-use in papers and presentations. Official website Human CNV browser Download data
Human genome replication timing
DNA replication creates opportunities for mutation, and the timing of DNA replication correlates with the density of SNPs across the human genome. To enable deeper investigation of how DNA replication timing relates to human mutation and variation, we generated a high-resolution map of the human genome’s replication timing program and analyzed its relationship to point mutations, copy number variations, and the meiotic recombination hotspots utilized by males and females. Amnon Koren makes available these data resources, which were generated and/or further analyzed in work he described in the following papers. These data are from Koren’s experiments profiling replication timing at loci across the human genome in lymphoblastoid cell lines. The experimental protocol was developed by Amnon Koren and is described in the AJHG paper. Download data
Birdsuite
Birdsuite is an open-source set of software tools for analyzing data from SNP arrays to detect and report SNP genotypes, common copy-number polymorphisms (CNPs), and novel, rare, or de novo copy-number variants (CNVs). Birdsuite analyzes data generated using “hybrid” SNP/CNV genotyping arrays (e.g., the SNP 6.0 array) that we co-developed with Affymetrix. While most of the components of the suite can be run individually (for instance, for SNP genotyping only), the Birdsuite is especially intended for integrated analysis of SNPs and CNVs. The two papers describing hybrid SNP/CNV arrays and Birdsuite have been cited respectively in 712 and 529 academic papers as of February 2015. Official Birdsuite website.
Drop-seq
Drop-seq is a technology we developed to enable biologists to analyze genome-wide gene expression in thousands of individual cells in a single experiment. Our Drop-seq resources site provides interested users with resources to implement Drop-seq in their own labs. We hope Drop-seq helps you do things you have always wanted to do. Tell us about it!
Drop-phase

Drop-phase is an approach we developed to quickly determine the chromosomal phase of sequence variants by separating genomic DNA into thousands of nanoliter droplets and then analyzing the extent to which alleles partition into the same or different droplets. We hope this is useful for studies involving genome editing, allele-specific expression, and for clinical scenarios involving compound heterozygosity.