2025

Long somatic DNA-repeat expansion drives neurodegeneration in Huntington’s disease
Robert E. Handsaker, Seva Kashin, Nora M. Reed, Steven Tan, Won-Seok Lee, Tara M. McDonald, Kiely Morris, Nolan Kamitaki, Christopher D. Mullally, Neda R. Morakabati, Melissa Goldman, Gabriel Lind, Rhea Kohli, Elisabeth Lawton, Marina Hogan, Kiku Ichihara, Sabina Berretta, Steven A. McCarroll
(Cell, January 2025)
Huntington Disease (HD) is a fatal genetic brain disorder in which most of a person’s striatal projection neurons (SPNs) degenerate and die. Science has long sought to understand why SPNs are so vulnerable in HD, why this pathology follows decades of apparent health, and how the disease-causing inherited DNA repeat (CAGn, n > 36) in the huntingtin (HTT) gene leads to this neurodegeneration. This DNA repeat exhibits somatic mosaicism (variable length); we developed a way to measure its length together with genome-wide RNA expression in the same individual cells. We found that, in persons with typical inherited HD-causing alleles (of < 50 CAG repeats), the CAG-repeat tract routinely expanded to 100-500+ CAG repeats in SPNs but rarely if ever did so in striatal interneurons or glia. Surprisingly, gene expression in these persons’ individual SPNs exhibited no apparent relationship to those SPNs’ CAG-repeat lengths across a wide range (36-150 repeats). In contrast, sparse SPNs with longer (150-500+) CAG repeats had profound gene-expression distortions which affected hundreds of genes, escalated alongside further repeat expansion, and culminated in widespread gene de-repression and expression of senescence/apoptosis genes. Our experiments, analyses, and simulations suggest that individual SPNs undergo decades of biologically quiet DNA repeat expansion, then asynchronously enter a brief toxicity phase before dying. We conclude that, at any moment in time, most SPNs in persons with HD actually have a benign (but somewhat unstable) huntingtin gene; and that HD is a DNA process for almost all of a neuron’s life.
2024

A sequence of SVA retrotransposon insertions in ASIP shaped human pigmentation
Nolan Kamitaki, Margaux L. A. Hujoel, Ronen E. Mukamel, Edward Gebara, Steven A. McCarroll, Po-Ru Loh
(Nature Genetics, August 2024)
We uncovered how “jumping genes,” known as retrotransposons, have played a surprising role in shaping human traits. While genome-wide association studies identified genetic variation at the ASIP gene as a key player in skin pigmentation, the causal genetic variant remained elusive. We discovered that a family of SVA retrotransposons jumped into ASIP twice and influenced skin color and skin cancer risk. The first jump likely occurred before the separation of modern humans and Neanderthals and decreased ASIP’s abundance, leading to an increase in skin pigmentation. The second jump likely occurred in the ancestors of some modern Europeans and reversed the effect, leading to less pigmentation and increased skin cancer risk. The latter SVA insertion appears to be the functional link between the GWAS locus and skin traits, underscoring the importance of including structural variations in genetic studies to understand how DNA influences physical traits and disease.

Genetic drivers and cellular selection of female mosaic X chromosome loss
Aoxing Liu, Giulio Genovese, Yajie Zhao, Matti Pirinen, Seyedeh M. Zekavat, Katherine A. Kentistou, Zhiyu Yang, Kai Yu, Caitlyn Vlasschaert, Xiaoxi Liu, Derek W. Brown, Georgi Hudjashov, Bryan R. Gorman, Joe Dennis, Weiyin Zhou, Yukihide Momozawa, Saiju Pyarajan, Valdislav Tuzov, Fanny-Dhelia Pajuste, Mervi Aavikko, Timo P. Sipilä, Awaisa Ghazal, Wen-Yi Huang, Neal D. Freedman, Lei Song, Eugene J. Gardner, FinnGen, Estonian Biobank Research Team, Breast Cancer Association Consortium, Million Veteran Program, Vijay G. Sankaran, Aarno Palotie, Hanna M. Ollila, Taru Tukiainen, Stephen J. Chanock, Reedik Mägi, Pradeep Natarajan, Mark J. Daly, Alexander Bick, Steven A. McCarroll, Chikashi Terao, Po-Ru Loh, Andrea Ganna, John R. B. Perry, Mitchell J. Machiela
(Nature, June 2024)
In this study, we explored the genetic causes and health implications of mosaic loss of the X chromosome (mLOX) in females. We analyzed data from over 880,000 females from eight biobanks and found that 12% of females had detectable levels of mLOX in their white blood cells. To overcome the challenging problem of analyzing data from multiple biobanks each siloed in different computational frameworks we developed MoChA WDL, a minimalist and scalable set of pipelines to detect mosaic chromosomal alterations and run genome-wide association studies. We identified 56 common germline variants associated with mLOX, implicating genes related to chromosomal missegregation, cancer predisposition, and autoimmune diseases. Females with mLOX had an elevated risk of myeloid and lymphoid leukemias. This research highlights the importance of understanding mLOX to better understand the genetic factors that influence female health.
PMID: 38867047
Download PDF
X-planations: paper; MoChA WDL tool

Sibling chimerism among microglia in marmosets
(eLife, March 2024)
Chimerism is a biological phenomenon in which an organism contains cells from different organisms. While rare in most mammals, chimerism is common in marmosets due to shared blood circulation in utero of dizygotic twins and trizygotic triplets. Here, del Rosario et al. performed single-cell RNA-seq on a variety of tissues and blood across several marmosets and quantified the amount of chimerism across cell types and tissues. Chimerism was only detected in blood-derived cell types, answering a longstanding question in the field. In the brain, microglia and macrophages had abundant chimerism, with 18-64% of a marmoset’s microglia and macrophages derived from their birth sibling(s), and this level varied across brain regions. This work was featured in a recent eLife commentary by Chiou and Snyder-Mackler.
eLife Reviewed Preprint
Download PDF
bioRxiv page

A concerted neuron–astrocyte program declines in ageing and schizophrenia
(Nature, March 2024)
We discovered a relationship between neurons and astrocytes that we call the Synaptic Neuron-Astrocyte Program (SNAP), in which neurons and astrocytes coordinate gene expression related to synapses. We found that expression of SNAP varies across people and declines with advancing age and in persons with schizophrenia. Analysis of the genes recruited by SNAP in each cell type implicates astrocytes as well as neurons in shaping genetic risk for schizophrenia. We discovered SNAP by developing new computational ways to analyze single-nucleus RNA-seq data that we had generated from 191 brain donors. Our analyses suggest there is a shared biological basis for cognitive impairment in aging and schizophrenia, and illustrate how inter-individual variation can be used to reveal novel and surprising aspects of human brain biology.

BCFtools/liftover: an accurate and comprehensive tool to convert genetic variants across genome assemblies
(Bioinformatics, January 2024)
Researchers are often faced with updating genetic variants from old genome assemblies to newer genome assemblies. Current tools for this task have limitations which lead to the loss or incorrect conversion of genetic variants. Here, we introduce BCFtools/liftover, a tool designed to efficiently convert genomic coordinates with improved support for indels, single nucleotide variants and multi-allelic variants. Notably, BCFtools/liftover minimizes variant loss and is 10X faster than other tools, making it particularly useful for large-scale data conversions.
PMID: 38261650
Download PDF
X-planation
2023

A marmoset brain cell census reveals regional specialization of cellular identities
Fenna M. Krienen, Kirsten M. Levandowski, Heather Zaniewski, Ricardo C.H. Del Rosario, Margaret E. Schroeder, Melissa Goldman, Martin Wienisch, Alyssa Lutservitz, Victoria F. Beja-Glasser, Cindy Chen, Qiangge Zhang, Ken Y. Chan, Katelyn X. Li, Jitendra Sharma, Dana McCormack, Tay Won Shin, Andrew Harrahill, Eric Nyase, Gagandeep Mudhar, Abigail Mauermann, Alec Wysoker, James Nemesh, Seva Kashin, Josselyn Vergara, Gabriele Chelini, Jordane Dimidschstein, Sabina Berretta, Benjamin E. Deverman, Ed Boyden, Steven A. McCarroll, Guoping Feng
(Science Advances, October 2023)
Using single-nucleus RNA sequencing of over 2.4 million brain cells sampled from 16 locations in a primate (the common marmoset), we find that primate neurons are primarily imprinted by their region of origin, more so than by their functional identity.

Repeat polymorphisms underlie top genetic risk loci for glaucoma and colorectal cancer
(Cell, August 2023)
Using whole-genome sequencing data from >418,000 unrelated UK Biobank participants and >800 GTEx participants, we imputed variable numbers of tandem repeat (VNTR) lengths genome-wide to asses the role of VNTRs in complex traits and gene expression. Hundreds of VNRTRs were associated with complex traits and gene expression, including two non-coding VNTRs at TMCO1 and EIF3H that produce the largest contribution of known common genetic variation to risk of glaucoma and colorectal cancer, respectively.

Schizophrenia-associated somatic copy-number variants from 12,834 cases reveal recurrent NRXN1 and ABCB11 disruptions
(Cell Genomics, August 2023)
We detected somatic copy-number variants (sCNVs) in SNP-array data from >12,000 donors with schizophrenia (SCZ) and >11,000 controls from the Psychiatric Genomic Consortium. sCNVs were more common in donors with SCZ than controls. Additionally, recurrent sCNVs in NRXN1 and ABCB11 were observed in donors with SCZ. These results suggest that sCNVs may play a role in schizophrenia etiology.
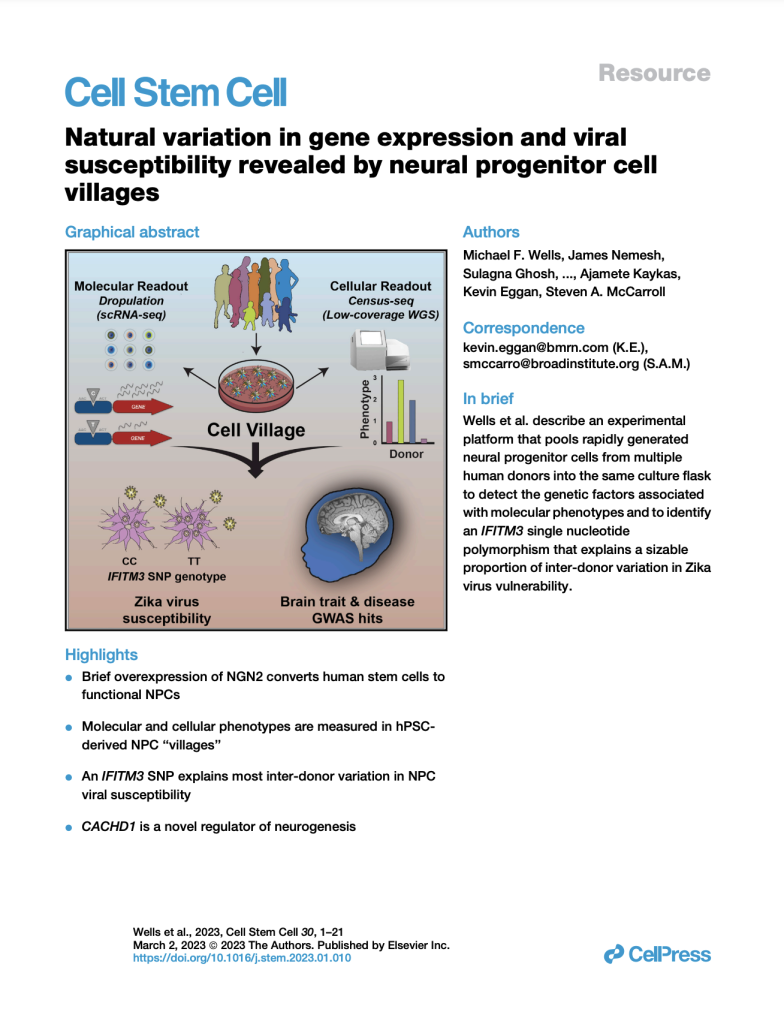
Natural variation in gene expression and viral susceptibility revealed by neural progenitor cell villages
(Cell Stem Cell, February 2023)
We developed a “cell village” experimental platform to analyze the genetic, molecular, and phenotypic heterogeneity across neural progenitor cells from 44 human donors cultured in a shared in vitro environment. To deconvolute the pooled signal, we developed Dropulation to assign cells to donors and Census-seq to assign cellular phenotypes to donors. This work uncovered a common IFITM3 SNP that explains the majority of the variation in Zika virus infectivity across donors.
PMID: 36796362
Download PDF
bioRxiv page
X-planation
2022

Ascertaining cells’ synaptic connections and RNA expression simultaneously with barcoded rabies virus libraries
Arpiar Saunders, Kee Wui Huang, Cassandra Vondrak, Christina Hughes, Karina Smolyar, Harsha Sen, Adrienne C. Philson, James Nemesh, Alec Wysoker, Seva Kashin, Bernardo L. Sabatini, Steven A. McCarroll
(Nature Communications, November 2022)
We introduce SBARRO (Synaptic Barcode Analysis by Retrograde Rabies ReadOut), a method that uses single-cell RNA sequencing to reveal directional, monosynaptic relationships based on the paths of a barcoded rabies virus from its “starter” postsynaptic cell to that cell’s presynaptic partners.

Chromosomal phase improves aneuploidy detection in non-invasive prenatal testing at low fetal DNA fractions
Giulio Genovese, Curtis J. Mello, Po-Ru Loh, Robert E. Handsaker, Seva Kashin, Christopher W. Whelan, Lucy A. Bayer-Zwirello, Steven A. McCarroll
(Scientific Reports, July 2022)
We present an approach that leverages the arrangement of alleles along homologous chromosomes—also known as chromosomal phase—to make non-invasive prenatal testing analyses more conclusive.

Whole-genome analysis of human embryonic stem cells enables rational line selection based on genetic variation
Florian T. Merkle, Sulagna Ghosh, Giulio Genovese, Robert E. Handsaker, Seva Kashin, Daniel Meyer, Konrad J Karczewski, Colm O’Dushlaine, Carlos Pato, Michele Pato, Daniel G. MacArthur, Steven A. McCarroll, Kevin Eggan
(Cell Stem Cell, February 2022)
We performed whole-genome sequencing (WGS) of 143 hESC lines and annotated their single-nucleotide and structural genetic variants. As a resource to enable reproducible hESC research and safer translation, we provide a user-friendly WGS data portal and a data-driven scheme for cell line maintenance and selection.
2021

Protein-coding repeat polymorphisms strongly shape diverse human phenotypes
Ronen E. Mukamel, Robert E. Handsaker, Maxwell A. Sherman, Alison R. Barton, Yiming Zheng, Steven A. McCarroll, Po-Ru Loh
(Science, September 2021)
We developed methods to estimate VNTR lengths from whole-exome sequencing data and impute VNTR alleles into single-nucleotide polymorphism haplotypes.
2020

Innovations present in the primate interneuron repertoire.
Fenna M. Krienen, Melissa Goldman, Qiangge Zhang, Ricardo C.H. Del Rosario, Marta Florio, Robert Machold, Arpiar Saunders, Kirsten Levandowski, Heather Zaniewski, Benjamin Schuman, Carolyn Wu, Alyssa Lutservitz, Christopher D. Mullally, Nora Reed, Elizabeth Bien, Laura Bortolin, Marian Fernandez-Otero, Jessica D. Lin, Alec Wysoker, James Nemesh, David Kulp, Monika Burns, Victor Tkachev, Richard Smith, Christopher A. Walsh, Jordane Dimidschstein, Bernardo Rudy, Leslie S. Kean, Sabina Berretta, Gord Fishell, Guoping Feng, Steven A. McCarroll.
(Nature, September 2020)
We profiled the single-cell RNA expression of more than 188,000 interneurons from humans, macaques, marmosets, and mice to assess the modifications, specializations, and innovations to brain cell types that occurred along each lineage.

Insights into dispersed duplications and complex structural mutations from whole genome sequencing 706 families.
Christopher W. Whelan, Robert E. Handsaker, Giulio Genovese, Seva Kashin, Monkol Lek, Jason Hughes, Joshua McElwee, Michael Lenardo, Daniel MacArthur, Steven A. McCarroll
(bioRxiv, August 2020)
We describe a new way to find and characterize dispersed duplications and complex de novo structural variation by utilizing identity-by-descent (IBD) relationships between siblings together with high-precision measurements of segmental copy number.

Insights into variation in meiosis from 31,228 human sperm genomes.
Avery Davis Bell, Curtis J Mello, James Nemesh, Sara A Brumbaugh, Alec Wysoker, Steven A McCarroll
(Nature, July 2020)
We sequenced the genomes of 31,228 gametes from 20 sperm donors, identifying 813,122 crossovers, 787 aneuploid chromosomes, and unexpected genomic anomalies.

Monogenic and polygenic inheritance become instruments for clonal selection.
Po-Ru Loh, Giulio Genovese & Steven A. McCarroll.
(Nature, June 2020)
To identify genes and mutations that give selective advantage to mutant clones, we identified among 482,789 UK Biobank participants some 19,632 autosomal mosaic chromosomal alterations (mCAs), including deletions, duplications, and copy number-neutral loss of heterozygosity (CNN-LOH).

Mapping genetic effects on cellular phenotypes with “cell villages”
Jana M. Mitchell, James Nemesh, Sulagna Ghosh, Robert E. Handsaker, Curtis J. Mello, Daniel Meyer, Kavya Raghunathan, Heather de Rivera, Matt Tegtmeyer, Derek Hawes, Anna Neumann, Ralda Nehme, Kevin Eggan, Steven A. McCarroll
(bioRxiv, June 2020)
Here we describe Census-seq, a way to measure cellular phenotypes in cells from many people simultaneously.

Absolute quantification and degradation evaluation of SARS-CoV-2 RNA by droplet digital PCR
(medRxiv, June 2020)

Complement genes contribute sex-biased vulnerability in diverse disorders.
Nolan Kamitaki, Aswin Sekar, Robert E Handsaker, Heather de Rivera, Katherine Tooley, David L Morris, Kimberly E Taylor, Christopher W Whelan, Philip Tombleson, Loes M Olde Loohuis, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Michael Boehnke, Robert P Kimberly, Kenneth M Kaufman, John B Harley, Carl D Langefeld, Christine E Seidman, Michele T Pato, Carlos N Pato, Roel A Ophoff, Robert R Graham, Lindsey A Criswell, Timothy J Vyse, Steven A McCarroll.
(Nature, May 2020)
Here we show that the complement component 4 (C4) genes in the MHC locus generate 7-fold variation in risk for lupus and 16-fold variation in risk for Sjögren’s syndrome. The same alleles that increase risk for schizophrenia greatly reduced risk for lupus and Sjögren’s syndrome. In all three illnesses, C4 alleles acted more strongly in men than in women.
2019

Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes.
Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, Marsh SE, Saunders A, Macosko E, Ginhoux F, Chen J, Franklin RJM, Piao X, McCarroll SA, Stevens B.
(Immunity, 2019)
We analyzed the RNA expression patterns of more than 76,000 individual microglia in mice during development, in old age, and after brain injury.
2018

Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain.
Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA.
(Cell, 2018)
We unmask the unique genetic signatures of more than 560 cell populations across nine brain regions.

Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations.
Loh PR, Genovese G, Handsaker RE, Finucane HK, Reshef YA, Palamara PF, Birmann BM, Talkowski ME, Bakhoum SF, McCarroll SA, Price AL
(Nature, 2018)
We identify inherited and acquired mutations that drive a precancerous blood condition.


Analyzing Copy Number Variation with Droplet Digital PCR
Bell AD, Usher CL, McCarroll SA
(Methods in Molecular Biology, 2018)
We describe how we analyze copy number variants using ddPCR and review the design of effective assays, the performance of ddPCR with those assays, the optimization of reactions, and the interpretation of data.
2017

Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations
(Nature, 2017)

Cell diversity and network dynamics in photosensitive human brain organoids
(Nature, 2017)
2016

Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia
(Nature Neuroscience, 2016)

Recurring exon deletions in the HP (haptoglobin) gene contribute to lower blood cholesterol levels
(Nature Genetics, 2016)

Schizophrenia risk from complex variation of complement component 4
(Nature, 2016)
2015

Structural forms of the human amylase locus and their relationships to SNPs, haplotypes and obesity
Usher CL, Handsaker RE, Esko T, Tuke MA, Weedon MN, Hastie AR, Cao H, Moon JE, Kashin S, Fuchsberger C, Metspalu A, Pato CN, Pato MT, McCarthy MI, Boehnke M, Altshuler DM, Frayling TM, Hirschhorn JN, McCarroll SA.
(Nature Genetics, 2015)
We describe a way to analyze genomic regions of high structural complexity and apply it the human amylase locus, which encodes the enzymes that digest starch into sugar. Though this variation has been reported to be the human genome’s largest influence on obesity, we find that this is not the case.

Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets
(Cell, 2015)

Large multiallelic copy number variations in humans
Handsaker RE, Doren VV, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA.
(Nature Genetics, 2015)
We describe an intriguing form of a copy number variation, in which a gene or genetic locus is present in widely varying numbers of copies in different individuals.

A Rapid Molecular Approach for Chromosomal Phasing
Regan JF, Kamitaki N, Legler T, Cooper S, Klitgord N, Karlin-Neumann G, Wong C, Hodges S, Koehler R, Tzonev S, McCarroll SA.
(PLoS One, 2015)
We describe a molecular method for quickly determining the chromosomal phase of pairs of sequence variants, even when they are separated by hundreds of thousands of base pairs, by using droplets to isolate long chromosomal segments.
2014

Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence
Genovese G, Kähler AK, Handsaker R, Lindberg J, Rose SA, Bakhourn SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M, Höglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF, Sklar P, Grönberg H, Hultman CM, McCarroll SA.
(New England Journal of Medicine, 2014)
We describe a common pre-cancerous state, involving the clonal amplification of blood cells with somatic mutations, that is readily detected by DNA sequencing, is increasingly common as people age, and is associated with increased risk of blood cancer later in life.

Genetic Variation in Human DNA Replication Timing
Koren A, Handsaker RE, Kamitaki N, Karlić R, Ghosh S, Polak P, Eggan K, McCarroll SA.
(Cell, 2014)
We describe a new way to study DNA replication by using increasingly abundant whole genome sequence data, which we find contains signatures of DNA replication processes that were active in cells at the moment DNA was extracted from them. Using data from the 1000 Genomes Project, we find that aspects of genome replication vary from person to person and are controlled by genetic variation that affects the presence and utilization of replication origins.

Random replication of the inactive X chromosome
Koren A, McCarroll SA.
(Genome Research, 2014)
We find that DNA replication follows two strategies: slow, ordered replication associated with transcriptional activity, and rapid, unstructured, “random” replication of silent chromatin on the inactive X chromosome and the autosomes. The two strategies coexist int he same cell, yet are segregated in space and time.

Genome-scale neurogenetics: methodology and meaning
(Nature Neuroscience, 2014)
2013

Mapping the human reference genome’s missing sequence by three-way admixture in Latino genomes
Genovese G, Handsaker RE, Li H, Kenny EE, McCarroll SA.
(American Journal of Human Genetics, 2013)
We show that data from Latino genomes can be used to map a substantial fraction of the human genome’s remaining unmapped sequence.

Using population admixture to help complete maps of the human genome
Genovese G, Handsaker RE, Li H, Altemose N, Lindgren AM, Chambert K, Pasaniuc B, Price AL, Reich D, Morton CC, Pollak MR, Wilson JG, McCarroll SA.
(Nature Genetics, 2013)
We describe a way to map the human genome’s “missing pieces” – tens of megabases of apparently human genome sequence that had no home on maps of the human genome – by using mathematical patterns in the sequence variation that is present in admixed populations such as African Americans. Surprisingly, we find that much of this sequence has been hiding in and around the centromeres of human chromosomes.


Progress in the genetics of polygenic brain disorders: significant new challenges for neurobiology [Review Article]
McCarroll SA, Hyman SE.
(Neuron, 2013)
Advances in genome analysis are making possible successful genetic analyses of polygenic brain disorders. We outline the challenges and opportunities for neurobiology that lie ahead.

Our fallen genomes [Review Article]
Macosko EZ, McCarroll SA.
(Science, 2013)
Few human conceits are as relentlessly undermined by science as humans’ naïve assumptions about our own perfection. Charles Darwin abolished one such set of assumptions by showing that “inferior creations” are man’s evolutionary cousins. However, Darwin’s theory of evolution ultimately abetted a modern conceit—that the genomes in our cells are highly optimized end products of evolution.
2012

Exploring the variation within [Review Article]
Macoscko EZ, McCarroll SA.
(Nature Genetics, 2012)
We usually think of an individual’s cells as sharing the same genome.

Differential relationship of DNA replication timing to different forms of human mutation and variation
Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA.
(American Journal of Human Genetics, 2012)
We describe how DNA replication timing shapes the generation of new mutations across the human genome.

Structural haplotypes and recent evolution of the human 17q21.31 region
Boettger LM., Handsaker RE., Zody MC., McCarroll SA.
(Nature Genetics, 2012)
We describe an extreme form of structural variation at the human 17q21.31 inversion locus, which we find is segregating in at least nine different structural forms in human populations. We further show that complex genome structures can be analyzed by imputation from SNPs.
Before 2012

Discovery and genotyping of genome structural polymorphism by sequencing on a population scale
Handsaker RE, Korn JM, Nemesh J, McCarroll SA.
(Nature Genetics, 2011)
We describe a new class of methods for analyzing structural variation in whole genome sequence data.

Copy number variation and human genome maps [Review Article]
McCarroll SA.
(Nature Genetics, 2010)
Maps of human genome copy number variation (CNV) are maturing into useful resources for complex disease genetics.

Donor-recipient mismatch for common gene deletion polymorphisms in graft-versus-host disease.
McCarroll SA, Bradner JE, Turpeinen H, Volin L, Martin PJ, Chilewski SD, Antin JH, Lee SJ, Ruutu T, Storer B, Warren EH, Zhang B, Zhao LP, Ginsburg D, Soiffer RJ, Partanen J, Hansen JA, Ritz J, Palotie A, Altshuler D.
(Nature Genetics, 2009)
Transplantation and pregnancy, in which two diploid genomes reside in one body, can each lead to diseases in which immune cells from one individual target antigens encoded in the other’s genome. One such disease, graft-versus-host disease (GVHD) after hematopoietic stem cell transplantation (HSCT, or bone marrow transplant), is common even after transplants between HLA-identical siblings, indicating that cryptic histocompatibility loci exist outside the HLA locus. The immune system of an individual whose genome is homozygous for a gene deletion could recognize epitopes encoded by that gene as alloantigens. Analyzing common gene deletions in three HSCT cohorts (1,345 HLA-identical sibling donor-recipient pairs), we found that risk of acute GVHD was greater (odds ratio (OR) = 2.5; 95% confidence interval (CI) 1.4-4.6) when donor and recipient were mismatched for homozygous deletion of UGT2B17, a gene expressed in GVHD-affected tissues and giving rise to multiple histocompatibility antigens. Human genome structural variation merits investigation as a potential mechanism in diseases of alloimmunity.

Integrated detection and population-genetic analysis of SNPs and copy number variation.
McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wyoker A, Shapero MH, deBakker PIW, Maller J, Kirby A, Elliott AL, Parkin M, Hubbell E, Webster T, Mei R, Veitch J, Collins PJ, Handsaker R, Lincoln S, Nizzari MM, Blume J, Jones K, Rava R, Daly MJ, Gabriel SB, Altshuler DM.
(Nature Genetics, 2008)
Dissecting the genetic basis of disease risk requires measuring all forms of genetic variation, including SNPs and copy number variants (CNVs), and is enabled by accurate maps of their locations, frequencies and population-genetic properties. We designed a hybrid genotyping array (Affymetrix SNP 6.0) to simultaneously measure 906,600 SNPs and copy number at 1.8 million genomic locations. By characterizing 270 HapMap samples, we developed a map of human CNV (at 2-kb breakpoint resolution) informed by integer genotypes for 1,320 copy number polymorphisms (CNPs) that segregate at an allele frequency >1%. More than 80% of the sequence in previously reported CNV regions fell outside our estimated CNV boundaries, indicating that large (>100 kb) CNVs affect much less of the genome than initially reported. Approximately 80% of observed copy number differences between pairs of individuals were due to common CNPs with an allele frequency >5%, and more than 99% derived from inheritance rather than new mutation. Most common, diallelic CNPs were in strong linkage disequilibrium with SNPs, and most low-frequency CNVs segregated on specific SNP haplotypes.

Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease.
McCarroll SA, Huett AS, Kuballa P, Chilewski S, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ.
(Nature Genetics, 2008)
Following recent success in genome-wide association studies, a critical focus of human genetics is to understand how genetic variation at implicated loci influences cellular and disease processes. Crohn’s disease (CD) is associated with SNPs around IRGM, but coding-sequence variation has been excluded as a source of this association. We identified a common, 20-kb deletion polymorphism, immediately upstream of IRGM and in perfect linkage disequilibrium (r(2) = 1.0) with the most strongly CD-associated SNP, that causes IRGM to segregate in the population with two distinct upstream sequences. The deletion (CD risk) and reference (CD protective) haplotypes of IRGM showed distinct expression patterns. Manipulation of IRGM expression levels modulated cellular autophagy of internalized bacteria, a process implicated in CD. These results suggest that the CD association at IRGM arises from an alteration in IRGM regulation that affects the efficacy of autophagy and identify a common deletion polymorphism as a likely causal variant.

Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs.
Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, Lee C, Nizzari MM, Gabriel SB, Purcell S, Daly MJ, Altshuler D.
(Nature Genetics, 2008)
Accurate and complete measurement of single nucleotide (SNP) and copy number (CNV) variants, both common and rare, will be required to understand the role of genetic variation in disease. We present Birdsuite, a four-stage analytical framework instantiated in software for deriving integrated and mutually consistent copy number and SNP genotypes. The method sequentially assigns copy number across regions of common copy number polymorphisms (CNPs), calls genotypes of SNPs, identifies rare CNVs via a hidden Markov model (HMM), and generates an integrated sequence and copy number genotype at every locus (for example, including genotypes such as A-null, AAB and BBB in addition to AA, AB and BB calls). Such genotypes more accurately depict the underlying sequence of each individual, reducing the rate of apparent mendelian inconsistencies. The Birdsuite software is applied here to data from the Affymetrix SNP 6.0 array. Additionally, we describe a method, implemented in PLINK, to utilize these combined SNP and CNV genotypes for association testing with a phenotype.

Copy-number variation and association studies of human disease.
McCarroll SA, Altshuler DM.
(Nature Genetics, 2007)
The central goal of human genetics is to understand the inherited basis of human variation in phenotypes, elucidating human physiology, evolution and disease. Rare mutations have been found underlying two thousand mendelian diseases; more recently, it has become possible to assess systematically the contribution of common SNPs to complex disease. The known role of copy-number alterations in sporadic genomic disorders, combined with emerging information about inherited copy-number variation, indicate the importance of systematically assessing copy-number variants (CNVs), including common copy-number polymorphisms (CNPs), in disease. Here we discuss evidence that CNVs affect phenotypes, directions for basic knowledge to support clinical study of CNVs, the challenge of genotyping CNPs in clinical cohorts, the use of SNPs as markers for CNPs and statistical challenges in testing CNVs for association with disease. Critical needs are high-resolution maps of common CNPs and techniques that accurately determine the allelic state of affected individuals.

Common deletion polymorphisms in the human genome.
McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett J, Dallaire S, Gabriel SB, Lee C, Daly MJ, Altshuler DM.
(Nature Genetics, 2006)
The locations and properties of common deletion variants in the human genome are largely unknown. We describe a systematic method for using dense SNP genotype data to discover deletions and its application to data from the International HapMap Consortium to characterize and catalogue segregating deletion variants across the human genome. We identified 541 deletion variants (94% novel) ranging from 1 kb to 745 kb in size; 278 of these variants were observed in multiple, unrelated individuals, 120 in the homozygous state. The coding exons of ten expressed genes were found to be commonly deleted, including multiple genes with roles in sex steroid metabolism, olfaction and drug response. These common deletion polymorphisms typically represent ancestral mutations that are in linkage disequilibrium with nearby SNPs, meaning that their association to disease can often be evaluated in the course of SNP-based whole-genome association studies.